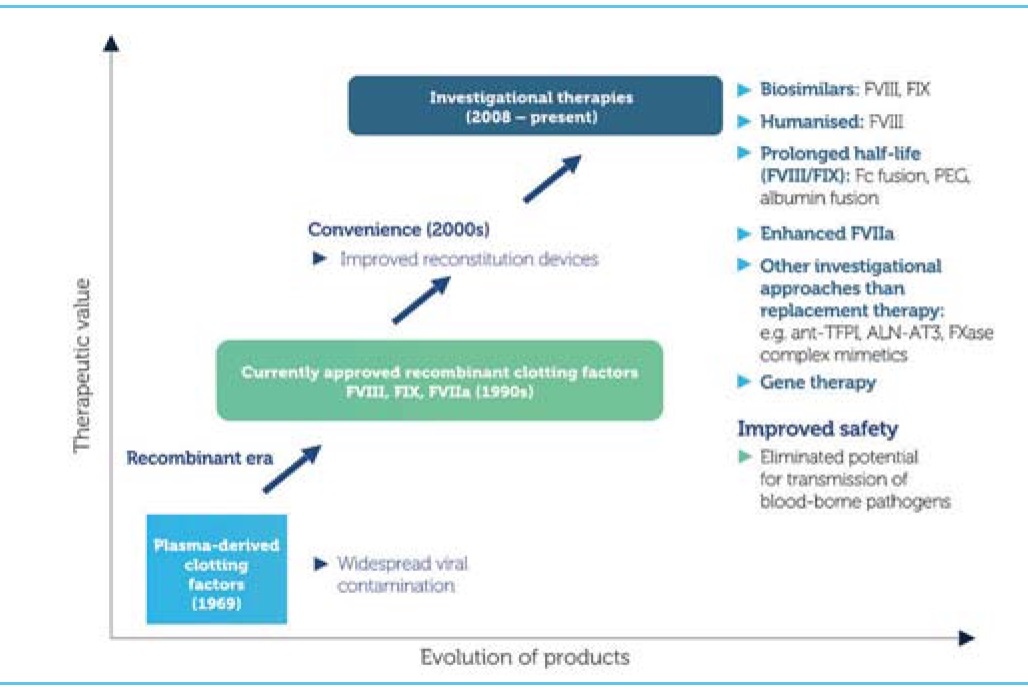
Haemophilia management has witnessed many milestones over the years. The first successful treatment of an inherited bleeding disorder with infused blood was back in 1840 [1]. The 1980s witnessed the purification and characterisation of factor IX [2] and factor VIII [3, 4, 5, 6], and the subsequent development of concentrates. The first recombinant factor VIII was produced in 1992 and the first recombinant factor IX was produced in 1997; this meant that patients could be treated with products free from contamination. Now there are novel therapies with the potential to transform the lives for individuals with inherited bleeding disorders and their families.
Improving clotting factor concentrates
Several strategies can be employed in order to improve the efficacy and extend the half-life of clotting factors. These include:
Prolonging efficacy by pegylation (the attachment of polyethylene glycol polymer chains to the clotting factor molecule, whether randomly or in a site-specific manner), hyperglycosylation (the addition of carbohydrate residues, an approach being used to extend the half-life of erythropoetin three- or fourfold), polysialation (the introduction of naturally occurring polymers to coat the protein, which has been shown to increase the half-life of factor VIII and reduce its clearance) and by the addition of fusion proteins, a recombinant technology in which factor is fused to the fragment crystallisation (Fc) portion of a natural molecule.
Higher potency clotting factors can be achieved by engineering proteins based on naturally occurring mutations with a higher specific activity. The best known example of this is the factor IX Padua mutation, which introduces a specific amino acid substitution into a factor IX protein, where following transfection into a muscle cell it has been shown to result in significant improvements in factor IX activity.
Clotting factors with reduced immunogenicity can also be made, which are much less likely to cause inhibitors. This may be achieved by expressing a human cell line with a glycosylation pattern that is more akin to the human protein than to one engineered in Chinese hamster ovary (CHO) cells. It is also possible to engineer the protein sequences, reducing, removing or altering those sequences that are particularly immunogenic. Hybrid porcine and human proteins can be made that remove the most immunogenic sequences, and recombinant porcine factor VIII will soon be available. It is also possible to generate factor IX that activates factor X independently of factor VIII, thereby removing the need for factor VIII.
Proteins that are resistant to inactivation can be made, either by stabilising the protein through the introduction of covalent bonds and modification of how the protein is broken down.
Improved production can be achieved using transgenic animals. The first transgenic recombinant protein was antithrombin synthesised in a goat and the antithrombin was isolated from the goat’s milk. It may even be possible to grow transgenic plants that produce clotting factors that can be absorbed orally.
The fusion protein approach is based on the neonatal Fc receptor, which permits the recycling of immunoglobulin G and to transport it across the placenta. Factor VIII or IX can be bound to the Fc portion of immunoglobulin G so that it becomes endocytosed by the endothelial cell, being protected within the lysosome and being recycled so it can disassociate at physiological pH when it passes back into the blood. This significantly prolongs the half-life of the protein.
The factor IX Fc fusion protein data indicate a significant prolongation of half-life, about three times longer than normal recombinant factor IX [7]. The factor VIII Fc fusion protein data is not quite as good, showing a prolongation of around 1.5 times longer than normal recombinant factor VIII [8]. This is probably due to the von Willebrand factor concentration, and unpublished data in von Willebrand Factor knockout mice suggest that half-life can be increased significantly more.
So, much can be done with clotting factors to increase their efficiency, to make them resistant to inhibitors, to reduce the risk of inhibitor formation and to improve their production. In addition, it has been shown that the half-life of factor VIII varies significantly between patients [9]. Therefore, dosing patients on the basis of their body weight and assuming a fixed half-life is illogical. With longer-acting products available clinicians will have to move more towards tailored dosing and treating patients as individuals.
Novel approaches
There are also some very exciting novel approaches that offer enormous hope for the future of haemophilia care, particularly for developing countries where clotting factor concentrates are expensive and access is problematic. But these approaches also offer great opportunities for those individuals with inhibitors who remain resistant to tolerisation. These approaches include modification of inhibition of coagulation by:
Additional approaches include:
Bispecific antibodies that simultaneously bind factors IXa and X and mimic the FVIIIa cofactor function [13]
Suppression of nonsense mutations by oral administration with ataluren, a molecule that is able to override premature stop codons to produce a full-length protein and is approved for Duchenne muscular dystrophy. Clinical trials are pending for this approach in haemophilia A and B due to nonsense mutations.
The literature is awash with novel approaches to treating haemophilia and other rare bleeding disorders, making this a very exciting time to be involved in haemostasis. Our ultimate aim, of course, is to improve the lives of individuals and their families with inherited bleeding disorders. A patient in his 50s who has had all of the problems associated with long-standing haemophilia recently said: “I’ve forgotten I have haemophilia. I can’t remember the last time I had a bleed.” This is a fantastic testament to how far we’ve come over the past 50 years.
David Perry
David Perry is Director of the Cambridge Haemophilia and Thrombophilia Centre, Consultant Haematologist & Associate Lecturer in Medicine in the Department of Haematology, Addenbrooke’s Hospital, Cambridge. He took up his present post approximately nine years ago and prior to this was Senior Lecturer/Honorary Consultant in Haemostasis and Thrombosis at the Royal Free Hospital, London. Dr Perry qualified from Edinburgh University Medical School and trained in haematology in Edinburgh and Birmingham. He was awarded a Wellcome Fellowship in Molecular Haematology in 1994 and spent 6 years with Professor RW Carrell in Cambridge investigating the molecular basis of antithrombin deficiency. Dr Perry has a major interest in medical education and in E-learning. In 2014 he was elected to Fellowship of the Academy of Medical Educators. Dr Perry currently chairs the NEQAS BC Steering Group, the NEQAS BC Specialist Advisory Group on Haemophilia Genetics, the UKHCDO working party on audit and various committees within the University of Cambridge.