The approach to inhibitor treatment and eradication at the University Hospital of Brussels is based on international data about inhibitor treatment. When a low titer (<5BU) inhibitor is detected, patients may continue to respond to FVIII replacement with minimal change in dose. Low titer inhibitors can be observed, as some are transient and will resolve spontaneously. A high titer inhibitor (> 5 BU) makes FVIII replacement ineffective and treatment of bleeding episodes requires the use of bypassing agents.
Currently available bypassing agents include recombinant activated factor VII (rFVIIa, NovoSeven, Novo Nordisk), and FEIBA (Baxter AG, Vienna, Austria). The rfVIIa facilitates hemostasis by activating factor X directly on the platelet surface thereby bypassing the tenase complex [3]. The half-life is 2.3 hours in adults but potentially shorter in children [4]. Attempts to engineer rFVIIa proteins with a longer circulation time are currently underway [5,6]. FEIBA (Factor Eight Inhibitor Bypassing Agent) induces thrombin generation and promotes haemostasis through a multifactorial mechanism of action that involves inhibitor bypassing activity in the coagulation pathway.FEIBA supplies prothrombin (also known as FII) and FXa, both critical components of the prothrombinase complex, where prothrombin is converted into thrombin [7].
The benefit of prophylactic therapy in haemophiliacs without an inhibitor has led many to consider prophylactic infusions of bypassing agents in haemophiliacs with an inhibitor [8]. Although some clinical trials with bypassing agents led to a reduction of bleeding frequency and improve health-related quality of life, it is not kown whether prophylaxis can improve joint health or reduce the rate of joint deterioration in untolerised inhibitor patients [9].
Immune tolerance (ITI) can be achieved in approximately 70% of patients [11,12]. Despite the development of multiple ITI protocols, the mechanism of tolerance induction and the best means to achieve it remains unknown.
Parameters influencing the success of ITI have been gained from several registries or from standard approaches in single institutions. In the International Immune Tolerance Registry (IITR), patients receiving ≥ 200IU/kg/day had the most favorable outcome, whereas the North American Immune Tolerance Registry (NAITR) found an inverse correlation between FVIII dose and success rate. However, lower doses of FVIII required a longer duration to achieve tolerance. Accordingly, the optimal dosing scheme of FVIII for ITI is unclear [13]. Optimal regimen (product or dose) for ITI remains to be defined. An International trial comparing 50 IU/kg three times a week to 200 IU/kg daily was recently stopped due to safety concerns (higher number of intercurrent bleeds) in the low-dose arm [10]. In both registries (NAITR and IITR) the pretreatment inhibitor titer (<10 BU) and the maximum historical titer (<200 BU) predicted successful ITI [11,12].
The type of FVIII product to use during ITI is also debatable [13,14]. Given the lack of compelling evidence for one product type over another, the product used before inhibitor formation was also used in the treatment of the inhibitor in the following two case studies.
In patients with a newly diagnosed inhibitor in which the inhibitor titer is higher than 10 BU, bypassing agents are used in the case of a bleed until the titer of the inhibitor is less than 10 BU before starting ITI.
Case Study 1 (Patient A)
Patient A was born in July 2004 and was diagnosed with severe haemophilia A aged 10 months when his general practitioner noticed a lot of bruises and asked for the blood to be examined. After the blood result (APTT 92 sec, n=33 sec) he referred patient A to our hematology unit.
The mother reported that she noticed bruising a few months after her child’s birth. These increased as the child became more active. She reported one episode of a swollen ankle, without any notion of trauma. Repeat blood tests revealed an APTT of 127 sec, and a FVIIIc <0.01 IU/dL, the diagnosis of haemophilia A was made. His genetic blood exam showed a mutation on exon 11 of FVIII gene (p.Cys573X).
He received six injections of recombinant FVIII for bleeding before we started prophylactic treatment (500 IU/once a week) on 13/04/2006. A month later, inhibitor development (84 BU/ml) was diagnosed after 10 doses of FVIII. FVIII was stopped at that point. There was a long discussion with the parents regarding the development and management of inhibitors. We decided to start ITI high dose regimen of 200 IU/kg/day when inhibitor level dropped <10BU/ml. Bleeds in this period were treated with rFVIIa 150μg/kg/2hr to avoid an anamnestic response.
In the period before the start of ITI, 25 bleeds were treated with rFVIIa. Before starting ITI, a port-a-cath (PAC) was inserted on 17/01/2008, when the inhibitor titer was 11BU/ml.
On 06/03/2008, the inhibitor titer was 4 BU/ml, we started ITI with high doses (200 IU/kg/day) of FVIII every day, (22 months after the appearance of the inhibitor). Figure 1 shows the value of the different titers of the inhibitor before the start of treatment. When ITI was started, the inhibitor titer increased due to amnestic response to FVIII exposure, the titer was >5000 BU (Figure 2). During ITI, the patient had several bleeds, mostly in his left ankle (target joint). These were successfully treated with rFVIIa. He commenced prophylactic rFVIIa in July 2008 due to repeated ankle bleeds; and as the inhibitor titer was 234 BU/ml. The prophylactic treatment was 2.4mg rFVIIa/day for almost one month, than every other day for a further two months. Afterwards one dose of 2.4 mg rFVIIa two times a week was given for a month. There were no bleeds in this period and the inhibitor level started also to decrease.
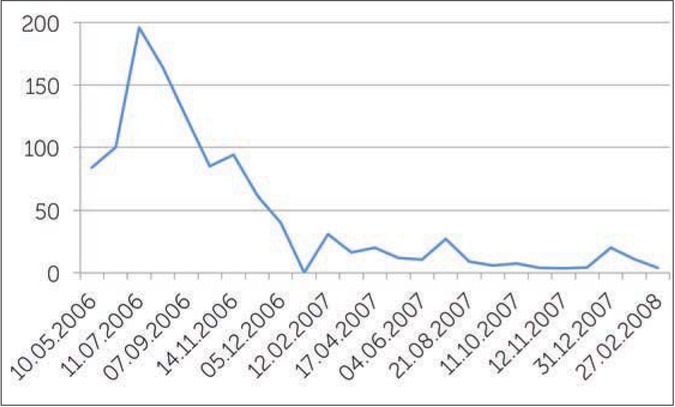
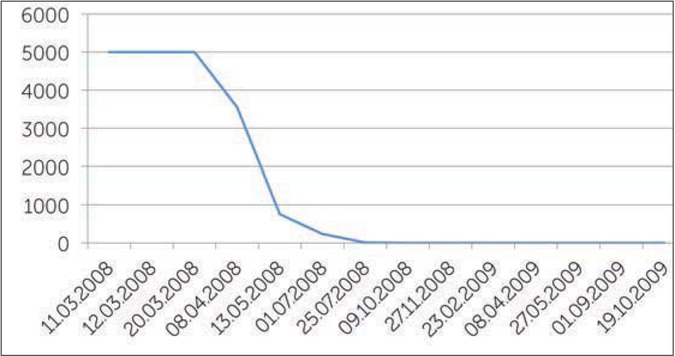
After almost one year the inhibitor level was 0.0 BU/ml. The results of the inhibitor remained negative and in August 2009 a recovery study of FVIII showed good recovery allowing a reduction in the dose of FVIII from 3000 IU FVIII/day (150 IU/kg/day) to 2000 IU rFVIII/day (100 IU/kg/day) for one week and then 1000 IU FVIII/day (50 IU/kg/day). The results of the inhibitor level remained negative and a further decrease in the amount of FVIII was continued.
At the end of September 2009 the patient was treated with 500 U/day (30 IU/kg/day). The inhibitor titer stayed negative. There were some concerns about the several bleeds he had in his left ankle. However, an MRI scan showed no abnormal structures of the bone, and the structure of the cartilage was normal. There was some liquid within the joint space (haemosiderin), but no thickening of the synovium [15]. Today, A is doing very well, and usually does his injections of 1000 U FVIII himself three times a week.
Case Study 2 (patient B)
Patient B was born in Romania where he was diagnosed with severe haemophilia A, with a FVIII plasma concentration of <2.5%. He arrived in our hospital in 2006 at the age of two years, with a few bruises and a haematoma on his forehead.
There was no family history of haemophilia. He received plasma derived FVIII “when needed”. There was no record of the amount of treatment he received in Romania, but the parents said that he did not receive very much because there were few bleeds other than some mouth bleeds and hematomas on the legs. However, communication with patient and parents was complex because of language difficulties.
The blood result in our hospital revealed revealed an APTT of 104 sec (n=33 sec), FVIII <0.16% and no inhibitor. His genetic blood exam showed an hemizygote delation of exons 8-12.
A new consultation was organized on the 28/09/2006. During that consultation the haematomas on both legs were still noticeable, but the haematoma on his forehead had gone. The treatment options were discussed with the parents;500 IU of FVIII (35 IU/kg) for minor bleeds, 50 IU/kg for life-threatening bleeds and as soon as treatment eligibility was organized to start prophylaxis.
Patient B was not seen again for more than a year, when he presented with haematoma of the gluteal muscle and scrotum. His parents said there had been no other issues apart from some bruising up until that time. He received three injections of FVIII (500 IU) for this bleeding and prophylaxis was proposed to start with a dose of 500 IU FVIII/week.
Prophylactic treatment was started on 10/09/2007 and at a consultation one month later we performed a blood exam and there was no inhibitor. There were no bruises; we decided to continue prophylactic treatment with 500 IU FVIII/week (difficulties to come to the hospital for injection). He had to come back in two months for follow-up and to visit the dentist for treatment of tooth cavities.
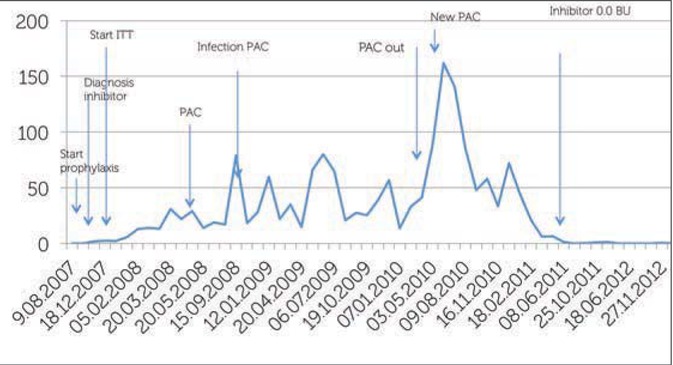
On 26/11/2007, we diagnosed an inhibitor after 14 infusions (11 prophylaxis – three for treatment of bleeds). The inhibitor level was 1.97 BU/ml and one month later was 2.44 BU/ml.
What was the treatment plan? We started ITI on 9/01/2008 with 50 IU of FVIII/kg twice a week (750 IU twice a week) and would manage bleeds with FVIIa 150 μg/kg/2h = 2.25 mg if necessary. The anamnestic response made the inhibitor increase to 12.9 BU/ml. Patient B had several bleeds, especially in his left elbow, during ITI and these were treated with rFVIIa. The inhibitor increased to 35 BU/ml, probably due to the bleeds.
A PAC was implanted to intensify the ITI treatment (three times a week 50 IU/kg of FVIII ), because of the increase of the inhibitor in May 2008). Because it was difficult for the patient to come to the hospital for his injections, we decided, after consultation with the parents, to organize home treatment for the injections three times a week.
Unfortunately, in September 2008, a PAC infection made his inhibitor increase to 80.0 BU/ml. The infection was treated with antibiotics (vancomycin via the PAC) and oxacillin (orally) with poor success. As a reaction to the increasing inhibitor, the doses of FVIII was increased to 100 IU/kg/day (2000 IU).
Due to several bleeds there was a limited extension of his elbow (-10%). Physiotherapists were charged to mobilize the joint with exercises. The inhibitor level was not really decreasing in that period, so the dose of FVIII was changed to 200 IU/kg/day. Several infections, like the tooth and PAC infection made it difficult to be successful with the inhibitor treatment.
In April 2010, the PAC was removed because of recurrent infections. The inhibitor titer was 162 BU. Because there was no PAC anymore, the FVIII dose was increased to 200 IU/kg five times per week via peripheral vein (4000 IU FVIII). He had to come to the unit for the injections. At that time, he went to the hospital school so it was easier to organize the injections. A new PAC was inserted in June 2010 allowing us to resume 200 IU FVIII/kg/day.
In August 2011, there was for the first time a negative inhibitor result, 3.5 years after the start of ITI low regimen and 19 months after the start of the high doses regimen. A recovery study was performed in October 2011 and again in November 2012 and the result showed a good recovery of FVIII. Patient B is now doing well with three injections of 1000 IU FVIII/week, there are no complications on his joints, the motion of the elbow is recovered. Figure 3 shows the level of inhibitor and also the most important interventions and changes in doses during ITI.
Conclusion
Inhibitor formation in patients with severe haemophilia has an enormous impact on the treatment of these patients. These two cases show that low-dose treatment may be associated with more bleeds (as seen in the international ITI study). Mostly, with the availability of good products, the bleeds can be treated, and if treated rapidly and adequately the outcome can be successful. The treatment of inhibitors is costly and requires a flexible approach but can have good outcomes.