Factor X (FX) deficiency is a rare autosomal recessive bleeding disorder with an incidence of 1:500,000 – 1:1,000,000, first described in 1905 [1]. The factor X gene is located on chromosome 13 (13q34), in the severe form with FX:C <0.01IU/ml bleeding results in high levels of morbidity and mortality. Severe FX deficiency is associated with spontaneous intracranial haemorrhage (ICH), gastrointestinal bleeding (GI) and haemarthrosis [2]. In type 1 deficiency both functional activity and antigen levels are proportionally decreased leading to severe bleeding manifestations, which usually present within the neonatal period [1].
Historically, treatment was given following bleeding episodes with fresh frozen plasma (FFP). However, the bleeding phenotype in factor X deficiency is severe and the concentration of factor X in FFP is low, and is associated with problems of volume overload when increasing levels into a therapeutic range, especially in children [3, 4].
FFP use has been superceded by prothrombin complex concentrates (PCC), which contain activated forms of factor II, VII, IX and X. Treatment is usually administered prophylactically in severe factor X deficiency, ensuring adequate FX trough levels to reduce the risk of bleeding. Factor X concentration in PCCs is inconsistent; there is a risk of thromboembolic complications given the presence of activated forms of other vitamin K-dependent factor concentrates [2,3].
BPL Factor X concentrate has been developed for use in treatment of hereditary factor X deficiency for both on-demand and regular prophylactic therapy as well as for surgical prophylaxis. Adult trials have recently been completed, with paediatric trials soon to start in order to obtain a product license [5]. BPL Factor X is made from plasma from donor-screened American subjects and undergoes three viral inactivation steps: solvent detergent, 15nm virus filtration and dry heat treatment. It contains a high factor X content of 100IU/ml, and is available in vials of 250IU and 500IU [SmPC].
We describe the compassionate named-patient use of BPL Factor X for prophylaxis in a nine-year-old girl with thromboembolic complications following replacement therapy with PCC.
Materials and methods
Presentation
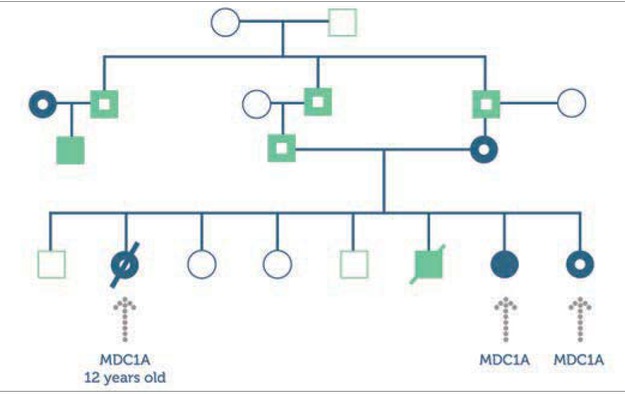
‘Sally’ (a pseudonym used for the purpose of this case report) is the seventh of eight children of consanguineous Caucasian parents. A previous child in this family died following intracranial haemorrhage (ICH) secondary to Factor X deficiency (Figure 1).
Sally was born in a maternity unit not affiliated with a haemophilia centre. At delivery, there were no bleeding issues in either Sally or her mother. Sally was not tested for factor X deficiency as her parents had failed to engage with local health care providers and had not disclosed their family history.
Sally was readmitted to hospital aged 6 days following blood streaked stools and umbilical cord bleeding. A diagnosis of severe FX deficiency was confirmed on coagulation screen (PT 25.8 (n8.2-14.1) APTT 71.1 (n=28-55) TT 17.2 (n=9.2-15.0) Fibrinogen 3.4g/L (n=1.7-4) and factor assays (Factor X <0.01IU/ml). FFP was administered prior to transfer to Great Ormond Street Hospital. Subsequent genetic analysis showed that Sally was homozygous for a missense mutation (Pro382Leu) within exon 8 in the factor X gene. Her parents are, as expected, both carriers of this mutation, as are several of her siblings. This is important information for their future reproductive choices as they are limited to marriage within their community.
On examination Sally appeared to be floppy with poor muscular tone in her legs. A provisional diagnosis of ICH was made: a CT scan revealed no bleeding; subsequent physiotherapy assessment revealed global hypotonia with reduced limb movement and hip contractures. A second significant diagnosis of congenital merosin deficient muscular dystrophy (MDC1A) was made. MDC1A is another rare (0.7:100,000) autosomal genetic condition. The gene for MDC1A is found on chromosome 6 and is responsible for laminin α2 production, which contributes to the protein merosin which is important in muscle development and strength. Children with MDC1A can be born with contractures, are unable to move their limbs independently, often develop scoliosis, require nutritional support via naso-gastric or gastrostomy feeding, and have a limited life expectancy with respiratory problems secondary to weakness of respiratory muscles often contributing to their demise [6]. It later emerged that Sally already had an older sibling with this condition who has since died. When she was 3 years of age another sister was also born with MDC1A (Figure 1).
Venous access was required to facilitate prophylactic therapy with PCC; a central venous access device (Hickman line) was inserted when Sally was 7 days old. PCC was given every day in the immediate post-operative period, decreasing to every 72 hours to maintain a trough factor X level of >0.10IU/ml. When Sally was 11 months old the Hickman line was converted to an indwelling venous access device (portacath). Due to recurrent infection, with numerous pathogens for which no source could be identified, a further three portacaths were placed over the following four years.
The fifth portacath (placed in 2008) lasted for 24 months, at which point it stopped bleeding back: thrombolytic therapy with TPA failed but it was still possible to infuse into the port. Eventually in 2010, the port occluded and split; PCC could no longer be infused intravenously, Sally was brought to hospital as an emergency with a painful swelling laterally to the port site where PCC had extravasated from the split tubing. Radiological imaging revealed occlusion of the right subclavian and right brachiocephalic veins with thrombus, and filling of the superior vena cava via collateral blood vessels. Sally’s factor X trough levels on PCC 72-hours post-dose were 13IU/ml; the thrombus was thought to be related to her immobility secondary to MDC1A and infusion of activated vitamin K dependent factors in the PCC.
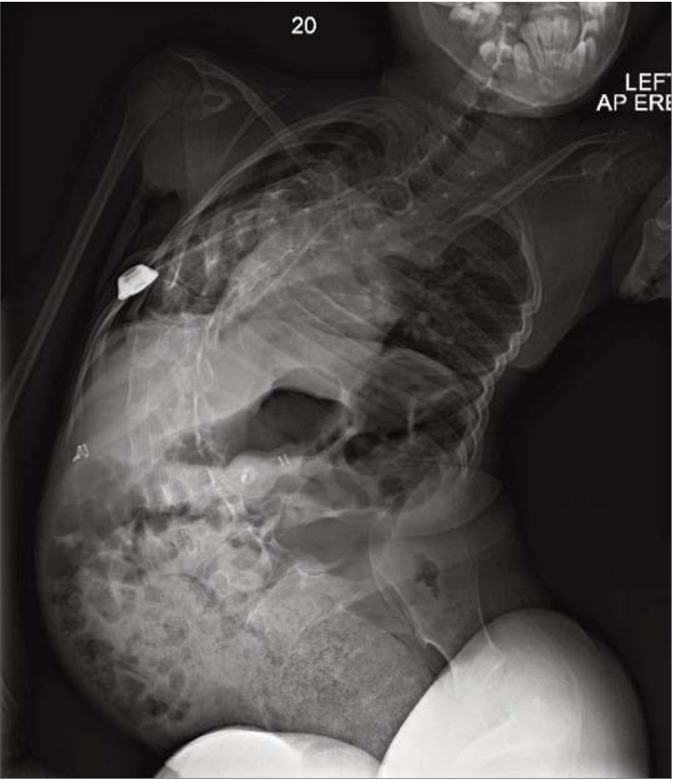
Portacath re-insertion by interventional radiologists failed. PCC prophylaxis was attempted peripherally but was complicated by journey times to and from hospital and became increasingly difficult over the following months. Limited venous access was exacerbated by positional complications of muscular dystrophy, she now had significant scoliosis (Figure 2) and elbow contractures, which made positioning for peripheral treatment virtually impossible, only the ulnar veins, were accessible. A clinical decision was made to treat her with BPL Factor X due to:
Figure 2
Complications including significant scoliosis complicated positioning for peripheral treatment
On-going difficulties with peripheral venous access
Lower infusion volumes in factor X compared to PCC
Previous thrombotic events related to PCC
Increasing immobility
Parental wish to continue prophylaxis, to reduce risk of intracranial haemorrhage.
Sally was commenced on BPL Factor X (50IU/Kg/dose) given peripherally twice-weekly maintaining a trough level of 6IU/ml 72-hours post-dose. This continued at the haemophilia centre for 3 months. Sally’s parents struggled with travelling to the centre, and a local hospital was unable to treat her, thus a decision was made to attempt another portacath insertion. This was achieved, via a complicated trans-hepatic approach, and facilitated home treatment by Sally’s parents.
Outcome
Sally’s parents have now treated her at home, with BPL Factor X for 3 years with no adverse complications: the portacath remains in situ with no issues with blood sampling or infusion. Sally has had no bleeding problems, no further thrombotic complications despite worsening of her muscular dystrophy, which continues to affect her mobility; she is now fully wheelchair bound, unable to swallow, is enterally fed and is totally dependent for all activities of daily living. Sally has been monitored for blood-borne viral infections before and during BPL FactorX treatment. No viral infections or abnormalities in liver function tests have been detected.
Conclusion
We have found BPL Factor X to be safe and effective for routine prophylaxis in this girl with severe factor X deficiency complicated by coexistent MDC1A. Compared with PCC, BPL Factor X offers consistent dosing, smaller infusion volumes and reduced thrombotic risk. This has enabled continued prophylactic home therapy to be administered for the past 3 years. Trough FX levels are now ~13IU/ml 72-hours post-dose, and no bleeding or thrombotic episodes have been seen. This has given great peace of mind to Sally’s parents: they know that Sally’s life expectancy remains extremely limited given her MDC1A but are reassured that her risk of life-threatening bleeding has been minimised. New clinical trials to confirm the safety and efficacy of BPL Factor X in children are planned for the near future.
