An ethnographic exploration of the everyday lives of PwH reveals key themes including what PwH consider as ‘normal’, how PwH adapt to cope with uncertainties around treatment, and the unique challenges encountered by PwH at different life stages.

Haemophilia is a genetic condition, primarily affecting males [1]. It is a lifelong condition that impairs the clotting ability of the blood, resulting in prolonged internal or external bleeding [2]. This bleeding occurs due to the lack of an essential blood-clotting factor [1]. The severity of the clotting factor deficiency is generally proportional to the frequency and severity of the haemorrhages that characterise this condition. These haemorrhages, or ‘bleeds’, can occur spontaneously or following trauma; they occur in the joints, muscles and soft tissues, resulting in damage, bruising or swelling, often accompanied by pain and physical restrictions [3], and in the absence of treatment they can cause crippling arthropathy [1]. Historically haemophilia has contributed to a shorter life expectancy, but due to treatment advances [3] including the availability of safe and effective blood coagulation treatment [2,4], life expectancy for people with haemophilia is now approaching that of the general male population [5], and life with the condition has drastically improved in recent generations.
Despite these encouraging developments in treatment in recent generations [6,7,8,9], people with haemophilia (PwH) still face many challenges related to their condition in their everyday lives. From a clinical standpoint the existing challenges have been well documented [8,10,11]. The prevalence of bleeds remains a problem, with approximately 60% of people with haemophilia who use prophylaxis still experiencing bleeds at least once a year [12]. Physical impairment in the form of reduced mobility is also still a major issue for ageing PwH, with approximately 60% of PwH above the age of 18 having problems with reduced mobility in their daily activities due to joint damage [13]. In addition to these physical challenges, the psychological challenges associated with haemophilia are also well documented [14,15,16,17], with some data estimating that 50% of PwH suffer from a psychological or psychiatric condition [18]. However, knowledge on the personal experience of these challenges in the daily lives of PwH is still relatively limited [3,8,19,20]. Indeed, several reviews of the existing psychosocial issues of haemophilia have claimed a need for studies exploring patient perspectives, which could help provide guidance on how to improve care [21,22]. This emphasis on the importance of integrating the psychosocial domain within routine care is also in line with recent research on care for other chronic diseases [23].
Ethnography is a qualitative research methodology in which researchers observe people in their own real-world setting, as opposed to clinical settings, with the aim of understanding how they manage their daily lives, what they value, and what barriers there are to achieving their goals [24]. The techniques of ethnography allow researchers to go beyond what people say, to see what they actually do in the context of their lives [25]. This methodology takes an ‘experience-near’ vantage point, closer to the daily lived experiences of PwH, in order to produce a richer picture of what it means to live with haemophilia today [26]. Situating the experiences of PwH in their socio-cultural contexts is also key in this study's ethnographic approach [27].
The aim of ’the ‘Living Well with Haemophilia’ ethnographic research study was to investigate the everyday life of PwH, including experiences related to their condition and treatment, their personal ways of managing the condition, and unmet support needs. The study covered a wide demographic sample of PwH in Europe, following PwH in five countries (Italy, Germany, Spain, UK and Ireland) in their daily lives, and observing interactions with friends, family, and health care professionals (HCPs).
METHODS
Fieldwork preparation
In order to frame the study and provide important background knowledge about haemophilia and its history, the researchers conducted semi-structured interviews with haemophilia experts before undertaking the fieldwork. The experts included a specialist nurse at a paediatric haemophilia treatment centre (co-author NM), a practising psychologist working with PwH (co-author ATO), a physiotherapist, an anthropologist, and a medical psychologist working within the area of haemophilia. The insights obtained defined the research themes for the field guide used during the fieldwork.
Eight researchers trained in ethnographic methods (including co-authors TH, AML and ABL) conducted the fieldwork. Each researcher entered the field with the ‘field guide’, which included an interview guide, exercise instructions, and all other vital fieldwork information. This field guide, along with the real-time sharing of field notes online, allowed researchers to align and collaborate effectively as data were collected across many countries simultaneously [28].
Study population
Researchers recruited PwH through patient organisations in each country. The recruitment criteria aimed for a representative sample of PwH, screening candidates by haemophilia type, disease severity, treatment regimen, presence of inhibitors, and age range (under 12, 13–18, 19–49, 50+). The researchers also recruited HCPs in each country for a mix of experience levels as well as representation of larger and smaller clinics.
Participant observation
Researchers planned to spend one to two days with each person with haemophilia (and their families). PwH were typically met at their homes and the researchers followed their everyday lives by, for example, visiting their favourite local hangouts and planned activities, speaking with family members and friends, and often participating in common activities such as sharing a meal together. The time spent with PwH by the researchers also involved observing treatment administration, clinic visits, and HCP consultations when possible. Furthermore, researchers undertook on-site observations in haemophilia treatment centres, including interactions between PwH and HCPs. On average, the researcher spent 8–12 hours with individual PwH and their families and two to six hours with HCPs.
Semi-structured interviews
Researchers conducted semi-structured interviews [29] with PwH and HCPs that focused on key interest areas defined in an interview guide, while also allowing interviewers to tailor their questions to the interview context/situation, and to the people being interviewed. The interview guide focused on the following predefined research themes: risks and fears, aspirations and limitations, identity, mastery and adaptation, and trust. Each of the five themes were explored through four different lenses: haemophilia in the home, haemophilia in the world, haemophilia in time, and treatment and care. During the interview sessions, researchers also took PwH through a range of exercises, such as mapping their history with haemophilia, word association, and support network mapping.
Data analysis
The researchers used a multi-tiered grounded theory approach to analyse the collected data [30]. As part of a bottom-up qualitative research design [31], cross-case analysis, clustering exercises, and challenge mapping were employed to detect patterns in the data. The researchers first used cross-case analysis to detect larger patterns in the material, during which every case was analysed in comparison with the other cases collected. The researchers then conducted a clustering exercise, wherein patterns evident across empirical material were clustered, analysed and theorised upon in order to derive and record general themes and trends in the data. The research team also conducted a challenge mapping exercise to record the disease-related challenges identified for each respondent. These challenges were then grouped by category, as well as by respondent profile (i.e. age group, disease severity, treatment regimen, reported adherence level). Statistics presented in this article are based on self-assessment by the respondents or are based on an analysis of respondent behaviour and/or statements.
Ethical considerations
The study followed the ethical standards outlined by the ICC/ESOMAR International Code on Market, Opinion and Social Research and Data Analytics [32], which sets out global standards for self-regulation for researchers and data analysts, as well as relevant national standards for participating countries [33,34,35,36]. Respondents signed GDPR-compliant consent forms and all interviews were conducted by trained researchers.
Given the highly personal nature of the data collected in this study, respondent privacy and anonymity were of high priority. As guaranteed in the GDPR-compliant consent form that each PwH and HCP signed, the researchers handled each respondent's personal data with the utmost care. In order to identify the different participants, while preserving confidentiality, each respondent in the study has been given a unique identification number. Quotes and cases are labelled with the respondent number and the respondent's age range, e.g. Respondent 1 (teenager). All potentially identifying information from respondent cases has been omitted. (Quotations from interviews in Spanish, German, Italian have been translated to English by the fully fluent field researchers who conducted the interviews.)
RESULTS
Study population
The researchers met with 42 people with haemophilia A and nine people with haemophilia B aged between 1.5 and 82 years (Table 1). Interviews often included the wider social ecology of each PwH, including friends, family, and caregivers. The researchers also interviewed 18 HCPs from seven HTCs. The HCPs interviewed were haematologists (n=10), nurses (n=4), physiotherapists (n=2), a dentist (n=1), and a medical assistant (n=1).
Table 1
Demographics of PwH in the study (n=51)
All information in the table was self-reported by study participants.
Over 500 hours of interviews were collected and analysed for this study. A summary of PwH statistics based on these interviews are shown in Table 2. The main findings of this study pointed to four themes:
Perceived ‘normality’
Treatment practices
Uncertainty and adaptation
Life stage-specific challenges.
Table 2
Summary of PwH statistics
| COURSE | N=51 | PERCENTAGE |
|---|---|---|
| PwH who described their lives as normal | 24 of 501 | 48% |
| PwH who reported regular bleeds and/or joint pain, despite self-reported adherence to treatment | 22 of 452 | 49% |
| PwH who had experienced at least one bleed within the last year | 32 of 51 | 63% |
| PwH who reported disease-related limitations in activity levels | 40 of 51 | 78% |
| PwH who reported disease-related challenges with travel | 17 of 51 | 33% |
| PwH who stated that they tended to entrust their HCP with key decisions | 39 of 501 | 78% |
| PwH who described difficulty translating their HCP's concept of protection and activities based on factor levels into everyday life | 26 of 501 | 52% |
| PwH who described trying to ignore their condition, who were on prophylaxis, and were often or sometimes non-adherent | 8 of 93 | 89% |
| PwH who pointed to family members, patient organisations and summer camps as sources of information they use | 20 of 501 | 40% |
| PwH who reported that they obtain information from five or more different sources | 15 of 501 | 30% |
Perceived ‘normality’ despite a life far from normal
1
The difficult history of haemophilia appeared ingrained in the collective memory of the haemophilia community. Several respondents described having heard daunting stories of the past from older relatives, or meeting older people with haemophilia and witnessing the effects of the challenges they have faced. One young man with haemophilia, Respondent 44 (20s), learned about the struggles of the older generations at a haemophilia patient society meeting:
‘I heard terrifying stories from the older fellas at the [meeting of the society] (...) The ’80s were scandalously bad.’
In the medical establishment, HCPs and pharmaceutical companies often refer to the years before treatment was widely available, or the HIV/HCV crisis. Patient organisations do the same, and in some countries still have the fight for justice regarding the HIV/HCV scandal as the focus of their efforts.
Many respondents appeared to consider their lives with haemophilia to be relatively burden-free compared to these difficult years in the past. Nearly half (48%) of PwH in the study described their lives as ‘normal’, yet researchers observed that they experienced frequent bleeds, pain, and multiple limitations causing them to hold back from leading the life they wanted to lead. To clarify, ‘normal’ is not used here to refer to a prescriptive definition of what constitutes a normatively correct way of living; rather, ‘normal life’ here is a term that researchers encountered in the field, used by respondents to refer to a life relatively unburdened by their disease. Respondent 28 (teenager), who was on crutches when interviewed, said he was leading a “normal life” and was not limited by his condition physically. However, when he recently had to run to catch a bus, he developed a major bleed leaving him on crutches. This perception of a ‘normal life’, i.e. the perception of one's life as relatively unburdened by haemophilia, was observed among nearly half of our respondents. However, it is important to note that some PwH with inhibitors, who either experienced increased treatment burden due to immune tolerance induction or shorter half-life medications, or who were failing to reach satisfactory factor levels, did not share this experience of perceived ‘normality’.
Researchers also observed how, for many respondents, the perception of and attempt to live a ‘normal life’ could have negative health consequences. For example, Respondent 6 (20s) described “just wanting to live a normal life” even though it has had major health consequences for him. He said that he prefers to have his condition “in the background” of his life, rather than at the front of his thoughts. He did not hold back from activities despite getting bleeds and he often forgot to take his treatment. As he said:
“for me, living with haemophilia means living a normal life and doing what I want – even if it means I had to put my foot in ice for a weekend.”
Despite the pervasive narrative of life for PwH being ‘normal’, nearly half (49%) of PwH in the study reported regular bleeds and/or joint pain, even with self-reported adherence to treatment, and 63% had experienced at least one bleed within the last year. In addition to bleeds and pain, researchers observed externally and self-imposed limitations experienced by PwH on a daily basis, with the most commonly reported challenge being limited activity levels (78%). Furthermore, 33% of the PwH in the study reported travel-related challenges. For example, Respondent 7 (50s), a middle-aged man, significantly limited his travels as his haemophilia meant that travel insurance was expensive, and he also feared not having access to good treatment abroad if something should happen.
Another common observation related to social exclusion. Respondent 25 (under 12) experienced being excluded in school from almost all activities by his teachers, and Respondent 23 (teenager) described how he had been rejected from enrolling into his local primary school some years previously when the personnel learned of his haemophilia. PwH also described limitations in career prospects. For example, Respondent 28 (teenager) described how he was restricted from applying for many types of jobs because he had been officially labelled as severely disabled. Respondent 16 (40s) described how he was not called to job interviews until he hid that he had haemophilia:
“Many people recommended me to disclose my status as severely disabled – when I didn’t, I was invited to interviews.”
PwH also described social isolation; for example, Respondent 31 (80s), an elderly participant, described having significant difficulties leaving his house because of bad joints, and as a result he often felt lonely.
Data from our study indicates that those who self-reported as non-adherent often also resisted identifying as a person with haemophilia. Of the nine respondents on prophylaxis who were often or sometimes non-adherent, eight (89%) described trying to ignore their disease, and two even described their non-adherence as a tool through which they could remain empowered in the face of their condition, not letting it define the way they lived their lives.
Current treatment approaches try to assure basic protection for PwH
2
For the respondents, current approaches to treatment were often coupled with holding back from certain activities due to their condition. The majority of PwH in the study (78%) refrained from activities they would like to pursue, due to their disease. Respondent 34 (under 12) expressed a strong desire to play football like his friends, but was not allowed to by his mother due to his condition. Respondent 19 (20s) described having to “be more careful as a child” than other children and know the things he “shouldn't do”:
The same pattern of self-restriction was also evident in his travel experiences: he refrained from undertaking longer travels, and his longest trips away from his home in Germany were to nearby countries.
On the other hand, some PwH would put themselves at risk by pursuing personal goals that went beyond those covered by their treatment regimen. Respondent 9 (30s) had decided to go against the advice from his physician by continuing to play hockey with his friends for over ten years, until he had to stop due to accelerating problems in his joints (ankle, knee and elbow). Despite having given up hockey, he still fought through the pain and accepted the consequences when playing with his daughter or when playing golf:
There seemed to be a difference in priorities between HCPs and PwH in relation to treatment objectives. One Spanish haematologist described this tendency from his point of view, suggesting that PwH consider treatment success as being unburdened by their disease in their activities and relationships, whereas haematologists, in his experience, primarily considered success in technical terms such as minimising bleeds, avoiding inhibitor development and securing adherence to treatment. He explained:
Similarly, a German haematologist described that success, to him, was when a person with haemophilia has an annual bleed rate of zero and is coping well with administering and adhering to treatment, whereas he considered one to two bleeds per year to be “good enough”. This basic protection (i.e. lower bleed rate, successful administration of treatment and adherence), rather than the pursuit of activities and personal goals, was a common metric of success in our observations of treatment practices.
PwH felt uncertain about their protection and adapted their behaviour
3
The majority of PwH in the study (78%) described entrusting their HCP with key treatment-related decisions. According to our data, HCPs carried a lot of weight for providing PwH with information about their condition, even going so far as to give patients their private phone numbers and urging them to reach out whenever they had questions. In addition to information from their HCPs, PwH in our study also looked to an average of 2.61 other sources for information. Forty per cent of PwH described looking to family, patient organisations and summer camps as sources of information about treatment. Several HCPs felt that PwH were often being misinformed outside of the clinic. Overall, all PwH highlighted their HCPs as their main source of information in relation to haemophilia treatment.
Despite these typically close relationships with HCPs and the use of additional sources of information, over half of the PwH in the study (52%) found it difficult to translate the information provided by HCPs into an understanding of what activities were appropriate in everyday life based on their level of protection. Rather, researchers found that many PwH ended up creating their own mental models and adaptations around protection. When these beliefs around protection were inaccurate, PwH described engaging in risky behaviour or being overly cautious in terms of activities, treatment practices and protection levels.
One example of how the understanding of protection levels by PwH could potentially be unsafe according to established clinical knowledge was described by Respondent 7 (50+), a middle-aged participant. His approach to protection was reactive rather than preventive: for him, protection was not about prevention of bleeds, but about immediate treatment after the fact. He was convinced that the key to avoiding serious bleeds was not regular prophylaxis, but rather immediate injections after a trauma had occurred. This meant that he would often perform activities, such as gardening, in daily life with very low protection levels. Even though this had led to bleeds in the past, he felt safe and did not hold back, instead thinking that if he felt a bleed coming on he could simply treat after the fact.
On the contrary, researchers observed in the families of Respondents 40 and 41 (both under 12) how a mental model of protection could lead to overly cautious behaviour. The boys had recently transitioned from standard half-life to extended half-life treatments, but the parents still did not consider them to be more protected on non-treatment days. Even though the parents were told by their HCPs that the boys’ new treatment would have a longer half-life, they still thought of all factor products as providing the same protection. This meant that the parents did not want the boys to play any sports on these days and actively encouraged them to play video games instead. Due to this model of protection, the boys’ exposure to social occasions was severely limited.
Treatment approaches were not always tailored to life-stage challenges
4
The PwH in our study faced specific challenges depending on their life-stage, without necessarily receiving care tailored to each stage. Our research identified four distinct phases that carry different challenges and anxieties to both people living with haemophilia and their caregivers (Figure 1).
Figure 1
Level of treatment burden, activity restriction, and joint problems across life-stages for PwH
The levels represented in this diagram are estimations based on an analysis of the disease-related challenges of respondents with haemophilia by age group. In the analysis, every patient challenge was recorded for each respondent, then grouped by challenge category (i.e. treatment burden, activity restriction, joint problems) and age group.
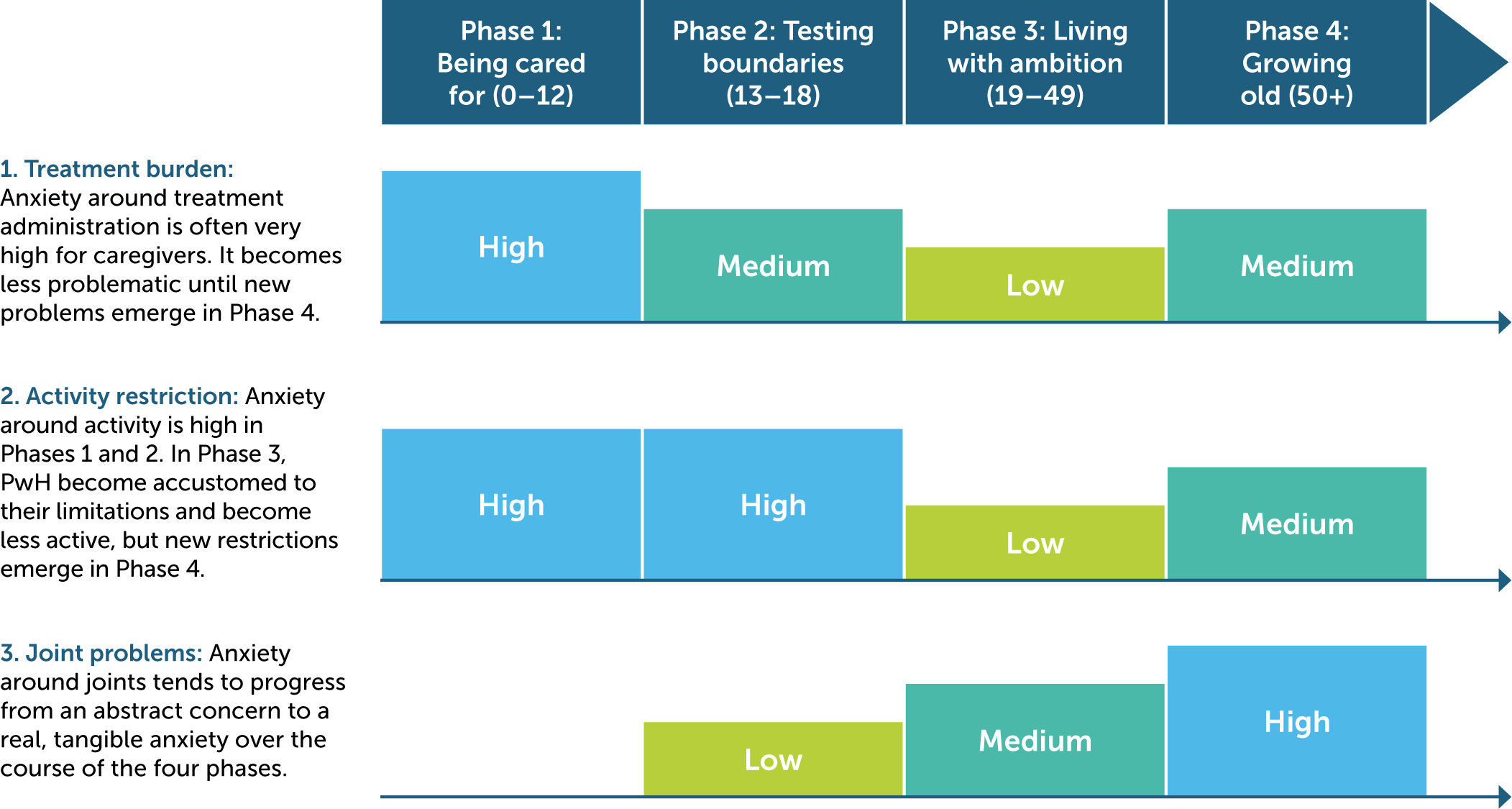
Phase 1: Being cared for
In the first phase, where the child with haemophilia was in the age range of 0–12 years old, most children lacked a developed embodied sense of their disease (i.e. difficulty assessing their limitations). Most caregivers described being focused on keeping the child safe and becoming accustomed to treatment administration as the main challenges, as well as feeling alone and lacking the necessary support that caring for a young child with haemophilia demands. Many parents described their greatest dilemma as being how to let their children test boundaries – to explore, fall and learn – while preserving their safety. The mother of Respondent 24 (under 12), a young boy, said:
The mother of Respondent 15 (under 12) said that only the sandbox was safe for her boy to play in – with everything else she was constantly worried.
Phase 2: Testing boundaries
In the second phase of challenges, the teenage years (13–19), PwH had often, but not always, learned to self-inject, and for that reason often described a lower treatment burden. For most teenage PwH, the challenge then was to fit in and not “feel like dust in the corner,” as Respondent 23 (teenager) described feeling when he was not able to do the same things as his friends in school. However, many teenagers in the study also rebelled against the limitations set by their condition by stretching their own limits. Respondent 2 (teenager) said:
Phase 3: Living with ambition
In the third phase, PwH aged 20–49 years, typically understood their (self-determined) limits and had settled into a certain lifestyle, which freed up mind space for other questions. In this phase, PwH tended to focus on building a career and a family, fighting through the pain to be able to live with ambition. In the family of Respondent 10 (30s), the daughter said:
In situations like this, treatment often fell into the background. As the father with haemophilia in this family said:
For most, anxieties around treatment and activities were at a low, but anxieties around joint problems were often beginning to emerge. In this group of participants, joint problems had progressed from an abstract concern to a present, tangible problem.
Phase 4: Growing old
In the fourth phase, researchers observed that PwH over 50 years of age in the study tried to embrace hobbies, but physical limitations and treatment problems often re-emerged, and joint problems became more all-encompassing. In most of the older PwH, the biggest challenge seemed to be staying mobile and finding ways to be able to do the things they wanted within the physical constraints of damaged joints from a life with haemophilia. Many experienced having to make trade-offs between being in pain and staying mobile, and it was challenging to stay active. For many, treatment had become difficult again due to joint problems or failing eyesight. Most elderly PwH had to face these problems on their own as there were so few before them that had reached an elderly age, as Respondent 31 (80s), an elderly man, said:
DISCUSSION
This study utilised ethnographic techniques of participant observation and semi-structured interviews to explore people's lives in their own unique contexts. The researchers employed this approach to studying life with haemophilia in an effort to produce a personal and contextual contribution to the existing body of knowledge on the challenges in the lives of PwH and how they may be addressed. Participant observation was an essential part of this approach, allowing researchers to come closer to an experience and understanding of the informants’ worlds from their point of view [37]. To gain insight about what it means to live with haemophilia today, researchers studied people living with haemophilia in their social context (their ecologies) and not as isolated individuals. This high level of contact allowed the research to go beyond surface level interactions to build a deeper understanding of the root cause of why people do what they do. Given the breadth of the study in terms of number of respondents [30], as well as its depth and explorative approach [3,38,39], the study was able to use qualitative analytic techniques, as well as quantitative techniques (e.g. basic statistical analysis). This unique approach aimed to both contribute to the existing body of knowledge on cataloguing and measuring challenges, as well as to the relatively limited existing body of knowledge on how these challenges play out in the daily lives of PwH.
The main findings of this study provide a picture of the ways in which PwH are prevented from living the fullest possible lives, despite ongoing improvements in care. The first main finding around perceived ‘normality’, explored the difference between what people say and how they actually live. Many PwH in the study described their lives as ‘normal’ (i.e. relatively free of haemophilia burden); however, by observing their everyday life, it was clear that these PwH were not leading a ‘normal life’ in terms of disease burden and experienced limitations. Another recent large-scale study also points to psychosocial challenges as well as physical challenges in PwH, with 50% experiencing constant pain in their daily lives [18]. The perspective of ‘normality’ could mean that PwH are more inclined to accept limitations that could potentially be addressed with treatment.
So why was this perspective of ‘normality’ so common? It seemed to stem from a belief deeply embedded in the haemophilia community that things used to be so much worse. This complicated combination of the ‘collective memory’ of a close-knit community that continues to echo the distressing experiences of PwH in the past (contamination of blood products, low availability of treatment, inadequate treatment options), and an appreciation of the medical advances of the past few decades may have led PwH to build a narrative of ‘normality’ around their experience. This discussion of ‘normality’ should not be confused with what Emiliani et al. (2011) refer to as ‘processes of normalization’, which actually refers to the integration of one's condition and care routine into one's daily life [39]. While Emiliani et al.'s ‘processes of normalization’ are seen to have potentially positive effects on patient outcomes and to be crucial for families adapting to the condition [36,40,41,42], a reluctance to acknowledge having haemophilia or the severity of one's condition has been shown to have negative effects on patient outcomes [43]. In fact, research points to non-adherence as one of the main explanatory variables in people's relationship with their disease [44].
The second main finding of the study suggests that while the current approaches to treatment allowed most PwH in the study to achieve a basic level of protection, it did not allow them to overcome the limitations on their daily life and personal goals imposed by their condition. The possible differences between the treatment goals of patients and HCPs have also been described in the literature [45]. HCPs often see prophylaxis treatment adherence as a goal in itself; however, from a patient perspective adherence to treatment recommendations is contingent on the adherence's potential for serving as a means for realising personal goals [6,46,47,48]. Recent research also points to a need for new measures of treatment success (e.g. patient-reported outcome measurement tools), as improvements in treatment have caused traditional metrics to become obsolete now that all treatments are seen as relatively successful. Recht et al. (2016) call for an increased level of personalisation for outcome measurement, whether clinical or patient-reported, that is capable of measuring ‘what matters most to patients’ [6]. This need for increased personalisation of care was also clear in the fourth main finding.
The third main finding of the study suggests that PwH developed their own mental models and individual care adaptations to navigate uncertainty around treatment, which can otherwise become a significant source of stress. HCPs and PwH were often misaligned in terms of their understanding of protection levels. From a clinical perspective, HCPs defined the window of opportunity for activities when patients are at a certain FVIII level. However, we found that for people living with haemophilia it was hard to understand how to practically implement this. When the information was unclear, many created personal ‘mental models’ and adapted the management of their disease to these models. Furthermore, existing research points also to PwH wanting more information from their HCPs that is easy to understand [49] and that can easily be translated into guidelines relevant to their personal situation, to be used to navigate the uncertainties they face in relation to their treatment.
The fourth main finding of the study suggests that PwH encounter unique challenges at four distinct age ranges and that many of these life-stage-specific challenges were unaddressed. Research has shown how other chronic conditions develop over a lifetime and can involve family members in attempts to overcome challenges [42,50]. The four phases defined here attempt to identify how challenges develop and change over the course of life with haemophilia. Limited treatment variation and personalisation between these phases were observed, despite clear differences in needs, aspirations, and activity levels. Although there is a need for further understanding of how ageing people with haemophilia cope with the onset of non-haemophilia related health issues [51] and how healthcare providers could address them [24], this study clearly suggests the potential for a more personalised approach to treatment.
LIMITATIONS OF THE STUDY
The findings described above are representative of patterns observed across several Western European countries. However, the sample size of this study was not sufficient to produce an analysis of country-specific differences within Europe. Further investigation is needed here.
CONCLUSION
The findings of the study suggest that a more personalised treatment approach could help to raise current standards of care and help to meet the needs of PwH in order for them to live a life without currently observed restrictions. A more personalised approach to care could address misplaced notions of ‘normality’ and uncertainty by allowing PwH to be more informed about their protection levels and how that should translate into their behaviour. An increasingly personalised approach to care could also ensure that PwH are protected for the activities and goals that are meaningful for them, beyond basic coverage. Similarly, it could ensure that the needs of PwH are met, even as those needs change across life stages. Such an approach to care could lead to better quality of life outcomes for PwH.
DISCLOSURES
This study was carried out by ReD Associates with funding and input from Sobi. Sobi and Sanofi reviewed the article. The authors had full editorial control of the article and provided their final approval of all content. TH, MBK, YG, AML, and ABL: Employees of ReD Associates. ATO: Researcher at the University of Muricia. NM: Researcher and Nurse at Alder Hey Children's Hospital. JS: Employee of Sobi.